
SKYRIZI 150 mg, solution injectable en stylo prérempli, boîte de 1 stylo prérempli de 1 mL
Dernière révision : 23/01/2025
Taux de TVA : 2.1%
Prix de vente : 2 481,83 €
Taux remboursement SS : 65%
Base remboursement SS : 2 481,83 €
Laboratoire exploitant : ABBVIE
Source :

Psoriasis en plaques
Skyrizi est indiqué dans le traitement du psoriasis en plaques modéré à sévère chez l'adulte qui nécessite un traitement systémique.
Rhumatisme psoriasique
Skyrizi, seul ou en association avec le méthotrexate (MTX), est indiqué dans le traitement du rhumatisme psoriasique actif chez l'adulte ayant présenté une réponse inadéquate ou une intolérance à un ou plusieurs traitements de fond antirhumatismaux (DMARD).
Hypersensibilité à la substance active ou à l'un des excipients mentionnés à la rubrique Liste des excipients.
Infections actives cliniquement importantes (par exemple : tuberculose active, voir rubrique Mises en garde spéciales et précautions d'emploi).
Traçabilité
Afin d'améliorer la traçabilité des médicaments biologiques, le nom et le numéro de lot du produit administré doivent être clairement enregistrés.
Infections
Le risankizumab peut augmenter le risque d'infection.
Chez les patients présentant une infection chronique, des antécédents d'infection récurrente, ou des facteurs de risque connus d'infections, le risankizumab doit être utilisé avec précaution. Le traitement par risankizumab ne doit pas être initié chez les patients présentant une infection active cliniquement importante tant que l'infection n'est pas guérie ou correctement traitée.
Les patients traités par risankizumab doivent être informés de la nécessité de consulter un médecin en cas de signes ou symptômes évocateurs d'une infection aiguë ou chronique cliniquement importante. En cas de développement d'une infection de ce type ou d'absence de réponse au traitement standard de l'infection, le patient doit être étroitement surveillé et le risankizumab ne doit pas être administré jusqu'à la guérison de l'infection.
Tuberculose
Un dépistage de la tuberculose (TB) doit être effectué préalablement à l'instauration du traitement par risankizumab. Les patients traités par risankizumab doivent être placés sous surveillance afin de rechercher les signes et symptômes de tuberculose active. Un traitement antituberculeux doit être envisagé avant le début du traitement par risankizumab chez les patients présentant des antécédents de tuberculose latente ou active et chez lesquels l'administration d'un traitement approprié ne peut être confirmée.
Vaccins
Avant l'initiation du traitement par risankizumab, l'administration de tous les vaccins nécessaires doit être envisagée conformément aux recommandations en vigueur en matière de vaccination. Si un patient a reçu un vaccin vivant (viral ou bactérien), il est recommandé d'attendre au moins 4 semaines avant de commencer le traitement par risankizumab. Les patients traités par risankizumab ne doivent pas recevoir de vaccins vivants pendant le traitement et au moins 21 semaines après l'arrêt du traitement (voir rubrique Propriétés pharmacocinétiques).
Hypersensibilité
Des réactions d'hypersensibilité graves, incluant l'anaphylaxie, ont été rapportées avec l'utilisation du risankizumab (voir rubrique Effets indésirables). En cas de survenue d'une réaction d'hypersensibilité grave, l'administration du risankizumab doit être immédiatement interrompue et un traitement approprié mis en œuvre.
Excipients à effet notoire
Skyrizi 150 mg, solution injectable en stylo prérempli ou seringue préremplie
Ce médicament contient moins de 1 mmol (23 mg) de sodium par stylo prérempli ou seringue préremplie, c'est-à-dire qu'il est essentiellement « sans sodium ».
Skyrizi 75 mg, solution injectable en seringue préremplie
Ce médicament contient 68,0 mg de sorbitol par dose de 150 mg.
L'effet additif des produits administrés concomitamment contenant du sorbitol (ou du fructose) et de l'apport alimentaire de sorbitol (ou de fructose) doit être pris en compte.
Ce médicament contient moins de 1 mmol (23 mg) de sodium par dose de 150 mg, c'est-à-dire qu'il est essentiellement « sans sodium ».
Résumé du profil de tolérance
Les effets indésirables les plus fréquemment rapportés ont été les infections des voies respiratoires supérieures (13,0 % dans le psoriasis).
Tableau récapitulatif des effets indésirables
Les effets indésirables du risankizumab observés lors des études cliniques (Tableau 1) sont répertoriés par classe de systèmes d'organes MedDRA selon la convention suivante : très fréquent (≥ 1/10) ; fréquent (≥ 1/100, < 1/10) ; peu fréquent (≥ 1/1 000, < 1/100) ; rare (≥ 1/10 000, < 1/1 000) ; très rare (< 1/10 000) et fréquence indéterminée (ne peut être estimée sur la base des données disponibles). Au sein de chaque groupe de fréquence, les effets indésirables sont présentés suivant un ordre de gravité décroissante.
Tableau 1 : Liste des effets indésirables
|
Classe de systèmes d'organes |
Fréquence |
Effets indésirables |
|
Infections et infestations |
Très fréquent |
Infection des voies respiratoires supérieuresa |
|
Fréquent |
Dermatophytosesb |
|
|
Peu fréquent |
Folliculite |
|
|
Affections du système immunitaire |
Rare |
Réactions anaphylactiques |
|
Affections du système nerveux |
Fréquent |
Céphaléec |
|
Affections de la peau et du tissu sous-cutané |
Fréquent |
Prurit Eruption cutanée Eczéma |
|
Peu fréquent |
Urticaire |
|
|
Troubles généraux et anomalies au site d'administration |
Fréquent |
Fatigued Réactions au site d'injectione |
|
a Inclut : infection de l'appareil respiratoire (virale, bactérienne ou non précisée), sinusite (notamment aiguë), rhinite, rhinopharyngite, pharyngite (notamment virale), angine, laryngite, trachéite. b Inclut : pied d'athlète, eczéma marginé de Hebra, dermatophytose de la peau glabre, pityriasis versicolor, tinea manuum, onychomycose, infection cutanée fongique. c
Inclut :
céphalée, céphalée de tension, céphalée d'origine sinusienne. d Inclut : fatigue, asthénie. e Inclut : au niveau du site d'injection : contusion, érythème, hématome, hémorragie, irritation, douleur, prurit, réaction, tuméfaction, induration, éruption cutanée. |
||
Description des effets indésirables sélectionnés
Infections
Le taux d'infections était de 75,5 évènements pour 100 patients-année pour les études cliniques dans le psoriasis et de 43,0 évènements pour 100 patients-année pour les études cliniques dans le rhumatisme psoriasique, incluant une exposition prolongée au risankizumab. La majorité des cas étaient non graves et d'intensité légère à modérée et n'ont pasconduit à l'arrêt du traitement par risankizumab. Le taux d'infections graves était de 1,7 évènement pour 100 patients-année pour les études cliniques dans le psoriasis et 2,6 évènements pour 100 patients-année pour les études cliniques dans le rhumatisme psoriasique (voir rubrique Mises en garde spéciales et précautions d'emploi).
Immunogénicité
Chez les patients traités par risankizumab à la dose clinique recommandée pendant une période pouvant aller jusqu'à 52 semaines lors des études cliniques dans le psoriasis, des anticorps antimédicament et anticorps neutralisants développés sous traitement ont été détectés chez respectivement 24 % (263/1 079) et 14 % (150/1 079) des patients évalués. Chez les patients exposés à un traitement de longue durée par le risankizumab dans l'étude d'extension, le profil d'immunogénicité observéjusqu'à la 204e semaine de traitement était similaire à celui des 52 premières semaines de traitement.
Chez la plupart des patients atteints de psoriasis, les anticorps anti-risankizumab (anticorps neutralisants inclus) n'ont pas été associés à des modifications de la réponse clinique ou de la tolérance. Parmi les quelques patients (approximativement 1 % ; 7/1 000 à la semaine 16 et 6/598 à la semaine 52) ayant des titres d'anticorps élevés (> 128), la réponse clinique semblait réduite. L'incidence des réactions au site d'injection était numériquement plus élevée dans le groupe positif pour les anticorps anti-médicament comparé au groupe négatif pour les anticorps anti-médicaments sur des périodes de traitement à court terme (à 16 semaines : 2,7 % vs 1,3 %) et à plus long terme (52 semaines : 5,0 % vs 3,3 %). Toutes les réactions au site d'injection étaient d'intensité légère à modérée, aucune n'a été grave et aucune n'a conduit à l'arrêt du traitement par risankizumab.
Chez les patients traités par risankizumab à la dose clinique recommandée pendant une période pouvant aller jusqu'à 28 semaines lors des études cliniques dans le rhumatisme psoriasique, des anticorps anti-médicament et des anticorps neutralisants ont été détectés chez respectivement 12,1 % (79/652) et 0 % (0/652) des patients étudiés. Les anticorps anti-risankizumab n'ont pas été associés à des modifications de la réponse clinique ou de la tolérance dans le rhumatisme psoriasique.
Rhumatisme psoriasique
Globalement, le profil de tolérance observé chez les patients atteints de rhumatisme psoriasiquetraités par risankizumab était similaire à celui observé chez les patients atteints de psoriasis en plaques.
Personnes âgées
Les données de tolérance sont limitées chez les sujets âgés de plus de 65 ans.
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via le système national de déclaration - voir Annexe V.
AVANT L'INSTAURATION du traitement :
- Effectuer un dépistage de la tuberculose.
- Envisager l'administration de tous les vaccins nécessaires
conformément aux recommandations en vigueur en matière de vaccination.
CONSULTER LE MEDECIN OU DEMANDER UNE ASSISTANCE MEDICALE DE TOUTE URGENCE en cas de :
- Signe de réaction allergique notamment :
• difficultés à respirer ou à avaler ;
• gonflement du visage, des lèvres, de la langue ou de la gorge ;
• tension artérielle basse, qui peut provoquer des vertiges ou des étourdissements
• démangeaisons sévères de la peau, accompagnées d'une éruption cutanée rouge ou de boutons.
- Symptômes d'une infection grave tels que :
• fièvre, symptômes pseudo-grippaux, sueurs nocturnes ;
• sensation de fatigue ou d'essoufflement, toux persistante ;
• peau chaude, rouge et douloureuse ou éruption cutanée douloureuse accompagnée de cloques.
INFORMER LE MEDECIN en cas de :
- infections des voies respiratoires supérieures accompagnées de symptômes tels que mal de gorge et nez bouché ;
- sensation de fatigue ; maux de tête ;
- mycose cutanée ;
- réactions au site d'injection (rougeur ou douleur) ;
- démangeaisons ; éruption cutanée ; eczéma ;
- apparition de petits boutons rouges sur la peau ; urticaire.
- vaccination récente ou à venir.
PATIENTES EN AGE DE PROCREER : Utiliser une contraception efficace
pendant le traitement et pendant au moins 21 semaines après l'arrêt du
traitement.
Femmes en âge de procréer
Les femmes en âge de procréer doivent utiliser une contraception efficace pendant le traitement et pendant au moins 21 semaines après l'arrêt du traitement.
Grossesse
Il n'existe pas de données ou il existe des données limitées (moins de 300 grossesses) sur l'utilisation du risankizumab chez la femme enceinte. Les études effectuées chez l'animal n'ont pas mis en évidence d'effets délétères directs ou indirects en ce qui concerne la toxicité sur la reproduction. Par mesure de précaution, il est préférable d'éviter l'utilisation du risankizumab pendant la grossesse.
Allaitement
On ne sait pas si le risankizumab est excrété dans le lait maternel. Les IgG humaines sont connues pour être excrétées dans le lait maternel durant les premiers jours qui suivent l'accouchement, l'excrétion diminuant jusqu'à de faibles concentrations peu après ; par conséquent, un risque pour le nourrisson allaité durant cette courte période ne peut être exclu. Une décision doit être prise d'interrompre le traitement / de s'abstenir de traiter par le risankizumab en prenant en compte le bénéfice de l'allaitement pour l'enfant au regard du bénéfice du traitement par risankizumab pour la femme.
Fertilité
L'effet du risankizumab sur la fertilité humaine n'a pas été évalué. Les études effectuées chez l'animal n'ont pas mis en évidence d'effets délétères directs ou indirects sur la fertilité.
Le risankizumab n'est pas supposé être métabolisé par les enzymes hépatiques ou éliminé par voie rénale. Aucune interaction n'est attendue entre le risankizumab et les inhibiteurs, inducteurs ou substrats des enzymes de métabolisation des médicaments ; par conséquent, aucune adaptation posologique n'est nécessaire (voir rubrique Propriétés pharmacocinétiques).
Traitement immunosuppresseur ou photothérapie concomitant(e)
La tolérance et l'efficacité du risankizumab en association avec les immunosuppresseurs, y compris les agents biologiques ou la photothérapie, n'ont pas été évaluées.
Ce médicament est destiné à être utilisé sous la conduite et la surveillance d'un médecin expérimenté dans le diagnostic et le traitement des pathologies pour lesquelles Skyrizi est indiqué.
Posologie
La dose recommandée est de 150 mg administrée en injection sous-cutanée à la semaine 0, à la semaine 4, puis toutes les 12 semaines (soit sous la forme de deux injections de 75 mg en seringues préremplies ou d'une injection de 150 mg en un stylo prérempli ou seringue préremplie).
Une interruption du traitement devra être envisagée chez les patients n'ayant pas répondu après 16 semaines de traitement. Chez certains patients atteints de psoriasis en plaques ayant obtenu une réponse initiale partielle, une amélioration ultérieure peut être observée en poursuivant le traitement au-delà de 16 semaines.
Oubli de dose
Si l'administration d'une dose a été oubliée, la dose doit être administrée dès que possible. L'administration doit ensuite reprendre selon le schéma habituel prévu.
Populations particulières
Personnes âgées
Aucune adaptation posologique n'est nécessaire (voir rubrique Propriétés pharmacocinétiques). Les données chez les sujets âgés de 65 ans et plus sont limitées.
Insuffisance rénale ou hépatique
Aucune étude spécifique n'a été menée pour évaluer l'effet de l'insuffisance hépatique ou rénale sur la pharmacocinétique du risankizumab. Ces affections ne sont généralement pas susceptibles d'avoir un impact significatif sur le profil pharmacocinétique des anticorps monoclonaux, aucune adaptation de la dose n'est jugée nécessaire (voir rubrique Propriétés pharmacocinétiques).
Population pédiatrique
La tolérance et l'efficacité du risankizumab chez les enfants et adolescents âgés de 5 à moins de 18 ans n'ont pas été établies. Aucune donnée n'est disponible.
Il n'existe pas d'utilisation justifiée du risankizumab chez les enfants âgés de moins de 6 ans dans l'indication du psoriasis en plaques modéré à sévère ou chez les enfants âgés de moins de 5 ans dans l'indication du rhumatisme psoriasique.
Patients en surpoids
Aucune adaptation posologique n'est nécessaire (voir rubrique Propriétés pharmacocinétiques).
Mode d'administration
Skyrizi est administré par injection sous-cutanée.
L'injection doit être administrée dans la cuisse ou l'abdomen. Les patients ne doivent pas s'injecter dans des zones où la peau est sensible, présente une ecchymose, un érythème, une induration ou des lésions psoriasiques.
Les patients peuvent s'injecter eux-mêmes Skyrizi après avoir été formés à la technique d'injection sous-cutanée. Les patients doivent être informés de la nécessité de lire les « Instructions d'utilisation » figurant dans la notice avant l'administration.
Seul un professionnel de santé ou un aidant peut administrer Skyrizi dans la partie supérieure externe du bras.
Skyrizi 75 mg, solution injectable en seringue préremplie
Le contenu des deux seringues préremplies doit être injecté afin d'administrer la dose complète de 150 mg. Les deux injections doivent être pratiquées dans des zones anatomiques différentes.
Durée de conservation :
2 ans
Précautions particulières de conservation :
À conserver au réfrigérateur (entre 2 °C et 8 °C). Ne
pas congeler.
Conserver le stylo prérempli dans l'emballage
extérieur à l'abri de la
lumière.
Le stylo prérempli de Skyrizi 150 mg peut être conservé en dehors du réfrigérateur (à une température ne dépassant pas 25 °C) pendant 24 heures au maximum dans l'emballage d'origine à l'abri de la lumière.
En l'absence d'études de compatibilité, ce médicament ne doit pas être mélangé avec d'autres médicaments.
En cas de surdosage, il est recommandé de surveiller le patient afin de détecter tout signe ou symptôme d'effet indésirable et d'initier immédiatement un traitement symptomatique approprié.
Classe pharmacothérapeutique : immunosuppresseurs, inhibiteurs d'interleukines, Code ATC : L04AC18
Mécanisme d'action
Le risankizumab est un anticorps monoclonal humanisé de type immunoglobuline G1 (IgG1) qui se lie sélectivement et avec une forte affinité à la sous-unité p19 de la cytokine humaine interleukine-23 (IL23) sans se lier à l'IL-12, ce qui inhibe l'interaction avec le complexe récepteur de l'IL-23.L'IL-23 est une cytokine impliquée dans les réponses immunitaires et inflammatoires. En empêchant la liaison de l'IL-23 à son récepteur, le risankizumab inhibe la voie de signalisation cellulaire de l'IL-23 et la libération des cytokines pro-inflammatoires.
Effets pharmacodynamiques
Lors d'une étude menée chez des patients atteints de psoriasis, l'expression des gènes associés à l'axe IL-23/IL-17 au niveau cutané a été diminuée suite à des doses uniques de risankizumab. Des réductions de l'épaisseur de l'épiderme, de l'infiltration des cellules inflammatoires et de l'expression des marqueurs de la pathologie psoriasique ont également été observées au niveau des lésions psoriasiques.
Lors d'une étude menée chez des patients atteints de rhumatisme psoriasique, une réduction statistiquement et cliniquement significative par rapport à l'inclusion a été observée à la semaine 24 pour les biomarqueurs associés à l'IL-23 et l'IL-17, notamment l'IL-17A, l'IL-17F et l'IL-22 plasmatiques, à la suite d'un traitement par risankizumab 150 mg par voie sous-cutanée à la semaine 0, à la semaine 4, puis toutes les 12 semaines.
Efficacité clinique et tolérance
Psoriasis en plaques
L'efficacité et la tolérance du risankizumab ont été évaluées chez 2 109 patients présentant un psoriasis en plaques modéré à sévère au cours de quatre études multicentriques randomisées en double aveugle (ULTIMMA-1, ULTIMMA-2, IMMHANCE et IMMVENT). Les patients inclus étaient âgés de 18 ans et plus et présentaient un psoriasis en plaques avec une surface corporelle atteinte (SCA) ≥ 10 %, un score sPGA (Static Physician Global Assessment) ≥3 dans l'évaluation globale du psoriasis (épaisseur/induration des plaques, érythèmes et desquamation) sur une échelle de sévérité de 0 à 4, un score PASI (Psoriasis Area and Severity Index) ≥ 12 et étaient candidats à un traitement systémique ou à la photothérapie.
Globalement, les patients présentaient à l'inclusion un score PASI médian de 17,8, une SCA médiane de 20,0 % et un score DLQI de 13,0. Le score sPGA initial était sévère chez 19,3 % des patients et modéré chez 80,7 % des patients. Au total, 9,8 % des patients de l'étude présentaient des antécédents de rhumatisme psoriasique diagnostiqué.
Sur l'ensemble des études, 30,9 % des patients étaient naïfs de tout traitement systémique (incluant traitements non biologiques et biologiques), 38,1 % avaient déjà reçu une photothérapie ou une puvathérapie, 48,3 % avaient déjà reçu un traitement systémique non biologique, 42,1 % avaient déjà reçu un traitement biologique et 23,7 % avaient reçu au moins un agent anti-TNF- alpha pour le traitement du psoriasis. Les patients ayant terminé ces études et d'autres études de phase 2/3 ont eu la possibilité de participer à une étude d'extension en ouvert, l'étude LIMMITLESS.
ULTIMMA-1 et ULTIMMA-2
Les études ULTIMMA-1 et ULTIMMA-2 ont inclus 997 patients (598 patients randomisés dans le groupe risankizumab 150 mg, 199 dans le groupe ustekinumab 45 mg ou 90 mg [en fonction du poids à l'inclusion] et 200 dans le groupe placebo). Les patients ont reçu le traitement à la semaine 0, à la semaine 4, puis toutes les 12 semaines. Dans ULTIMMA-1 et ULTIMMA-2, les deux co-critères principaux d'évaluation étaient la proportion de patients ayant obtenu 1) une réponse PASI 90 et 2) un score sPGA « blanchi » ou « minime » (sPGA 0 ou 1) à la semaine 16, versus placebo. Les résultats pour les co-critères principaux ainsi que pour les autres critères sont présentés dans le Tableau 2 et la Figure 1.
Tableau 2 : Résultats d'efficacité et de qualité de vie dans les études ULTIMMA-1 et ULTIMMA-2 chez des adultes atteints de psoriasis en plaques
|
|
ULTIMMA-1 |
ULTIMMA-2 |
||||
|
|
Risankizumab
(N = 304) n (%) | Ustekinumab
(N = 100) n (%) |
Placebo
(N = 102) n (%) |
Risankizumab
(N = 294) n (%) | Ustekinumab
(N = 99) n (%) |
Placebo
(N = 98) n (%) |
|
sPGA « blanchi « ou « minime » (0 ou 1) |
||||||
|
Semaine 16a |
267 (87,8) |
63 (63,0) |
8 (7,8) |
246 (83,7) |
61 (61,6) |
5 (5,1) |
|
Semaine 52 |
262 (86,2) |
54 (54,0) |
-- |
245 (83,3) |
54 (54,5) |
-- |
|
sPGA « blanchi » (0) |
||||||
|
Semaine 16 |
112 (36,8) |
14 (14,0) |
2 (2,0) |
150 (51,0) |
25 (25,3) |
3 (3,1) |
|
Semaine 52 |
175 (57,6) |
21 (21,0) |
-- |
175 (59,5) |
30 (30,3) |
-- |
|
PASI 75 |
||||||
|
Semaine 12 |
264 (86,8) |
70 (70,0) |
10 (9,8) |
261 (88,8) |
69 (69,7) |
8 (8,2) |
|
Semaine 52 |
279 (91,8) |
70 (70,0) |
-- |
269 (91,5) |
76 (76,8) |
-- |
|
PASI 90 |
||||||
|
Semaine 16a |
229 (75,3) |
42 (42,0) |
5 (4,9) |
220 (74,8) |
47 (47,5) |
2 (2,0) |
|
Semaine 52 |
249 (81,9) |
44 (44,0) |
-- |
237 (80,6) |
50 (50,5) |
-- |
|
PASI 100 |
||||||
|
Semaine 16 |
109 (35,9) |
12 (12,0) |
0 (0,0) |
149 (50,7) |
24 (24,2) |
2 (2,0) |
|
Semaine 52 |
171 (56,3) |
21 (21,0) |
-- |
175 (59,5) |
30 (30,3) |
-- |
|
DLQI 0 ou 1b |
||||||
|
Semaine 16 |
200 (65,8) |
43 (43,0) |
8 (7,8) |
196 (66,7) |
46 (46,5) |
4 (4,1) |
|
Semaine 52 |
229 (75,3) |
47 (47,0) |
- |
208 (70,7) |
44 (44,4) |
- |
|
PSS 0 (sans symptômes)c |
||||||
|
Semaine 16 |
89 (29,3) |
15 (15,0) |
2 (2,0) |
92 (31,3) |
15 (15,2) |
0 (0,0) |
|
Semaine 52 |
173 (56,9) |
30 (30,0) |
-- |
160 (54,4) |
30 (30,3) |
-- |
|
Toutes les comparaisons
du risankizumab versus ustekinumab et placebo ont obtenu une valeur p <
0,001, excepté pour le score PASI 75 à la semaine 52 dans ULTIMMA-2 avec une
valeur p = 0,001. a Co-critères
principaux d'évaluation versus placebo. b Pas d'impact sur la qualité de vie liée à la santé. c Échelle des symptômes du psoriasis (PSS), 0 signifiant l'absence des symptômes de douleur, démangeaisons, rougeur et brûlure au cours des dernières 24h. |
||||||
Figure 1 : Variation moyenne en pourcentage du score PASI au cours du temps par rapport à l'inclusion dans ULTIMMA-1 et ULTIMMA-2
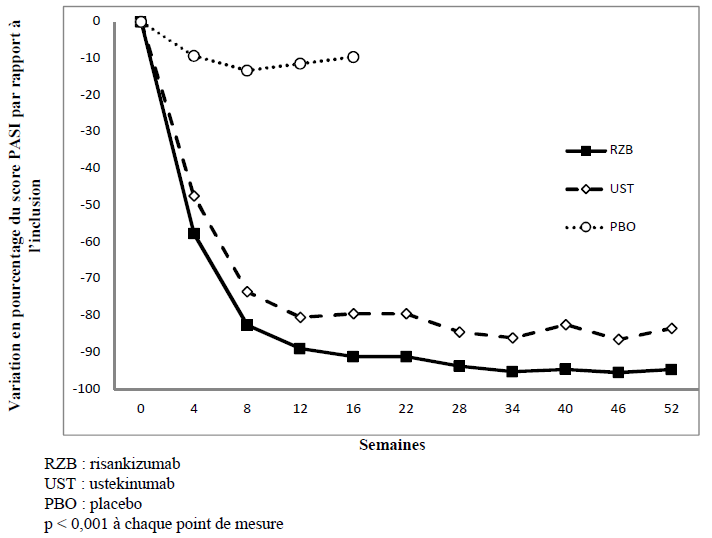
La prise en compte de l'âge, du sexe, de l'origine ethnique, du poids corporel ≤ 130 kg, du score PASI à l'inclusion, d'un rhumatisme psoriasique concomitant, de traitements systémiques non biologiques antérieurs, de traitements biologiques antérieurs et de précédents échecs d'une biothérapie n'a pas mis en évidence de différence dans la réponse à risankizumab selon les sous-groupes considérés.
Des améliorations du psoriasis ont été constatées, notamment au niveau du cuir chevelu, des ongles, de la paume de la main et de la plante des pieds à la semaine 16 et à la semaine 52 chez les patients traités par risankizumab.
Tableau 3 : Variations moyennes des scores NAPSI, PPASI, et PSSI par rapport à l'inclusion
|
|
ULTIMMA-1 |
ULTIMMA-2 |
IMMHANCE |
|||
|
|
Risankizumab |
Placebo |
Risankizumab |
Placebo |
Risankizumab |
Placebo |
|
NAPSI : variation à la semaine 16 (ES) |
N = 178 ; -9,0 (1,17) |
N = 56 ; 2,1 (1,86) *** |
N = 177 ; -7,5 (1,03) |
N = 49 ; 3,0 (1,76) *** |
N = 235 ; -7,5 (0,89) |
N = 58 ; 2,5 (1,70) *** |
|
PPASI : variation à la semaine 16 (ES) |
N = 95 ; -5,93 (0,324) |
N = 34 ; -3,17 (0,445) *** |
N = 86 ; -7,24 (0,558) |
N = 23 ; -3,74 (1,025) ** |
N = 113 ; -7,39 (0,654) |
N = 26 ; -0,27 (1,339) *** |
|
PSSI : variation à la semaine 16 (ES) |
N = 267 ; -17,6 (0,47) |
N = 92 ; -2,9 (0,69) *** |
N = 252 ; -18,4 (0,52) |
N = 83 ; -4,6 (0,82) *** |
N = 357 ; -20,1 (0,40) |
N = 88 ; -5,5 (0,77) *** |
|
NAPSI : variation à la semaine 52 (ES) |
N = 178 ; -15,7 (0,94) |
- |
N = 183 ; -16,7 (0,85) |
- |
- |
- |
|
PPASI : variation à la semaine 52 (ES) |
N = 95 ; -6,16 (0,296) |
- |
N = 89 ; -8,35 (0,274) |
- |
- |
- |
|
PSSI : variation à la semaine 52 (ES) |
N = 269 ; -17,9 (0,34) |
- |
N = 259 ; -18,8 (0,24) |
- |
- |
- |
|
Index de sévérité du psoriasis de l'ongle (NAPSI), Index de sévérité du psoriasis palmo-plantaire (PPASI), Index de sévérité du psoriasis du cuir chevelu (PSSI), et erreur standard (ES). ** P < 0,01 pour la comparaison avec le risankizumab. *** P < 0,001 pour la comparaison avec le risankizumab. |
||||||
L'anxiété et la dépression mesurées à l'aide de l'échelle HADS (Hospital Anxiety and Depression Scale) se sont améliorées dans le groupe risankizumab par rapport au groupe placebo à la semaine 16.
Maintien de la réponse
Dans une analyse combinée des patients recevant le risankizumab dans ULTIMMA-1 et ULTIMMA-2 concernant les répondeurs PASI 100 à la semaine 16, la réponse était maintenue à la semaine 52 chez 79,8 % (206/258) des patients ayant poursuivi le traitement par risankizumab. Concernant les répondeurs PASI 90 à la semaine 16, la réponse était maintenue à la semaine 52 chez 88,4 % (398/450) d'entre eux.
Chez les patients traités par risankizumab dans ULTIMMA-1 et ULTIMMA-2, 525 patients ont continué à recevoir le risankizumab toutes les 12 semaines dans LIMMITLESS. Parmi ces patients, 376 (71,6 %) ont terminé une période de traitement supplémentaire en ouvert de 252 semaines. Chez les patients ayant poursuivi l'étude, les améliorations des taux de réponse PASI 90 et de score sPGA « blanchi » ou « minime » obtenues avec le risankizumab à la semaine 52 ont été maintenues jusqu'à la semaine 304.
Chez les patients traités par ustekinumab dans ULTIMMA-1 et ULTIMMA-2, 172 patients ont reçu le risankizumab toutes les 12 semaines dans LIMMITLESS. Parmi ces patients, 116 (67,4 %) ont terminé l'étude, incluant la période de traitement par risankizumab en ouvert de 252 semaines et la période de suivi de fin d'étude. Chez les patients ayant poursuivi l'étude, les taux de réponse PASI 90 et de score sPGA « blanchi » ou « minime » ont augmenté de la semaine 52 à la semaine 76 et se sont ensuite maintenus jusqu'à la semaine 304.
Les Figures 2 et 3 présentent respectivement les taux de réponse PASI 90 et de score sPGA « blanchi » ou « minime », chez les patients ayant terminé 252 semaines de traitement en ouvert dans
LIMMITLESS.
Figure 2 : Pourcentage de patients ayant obtenu une réponse PASI 90 dans LIMMITLESS (cas observés)
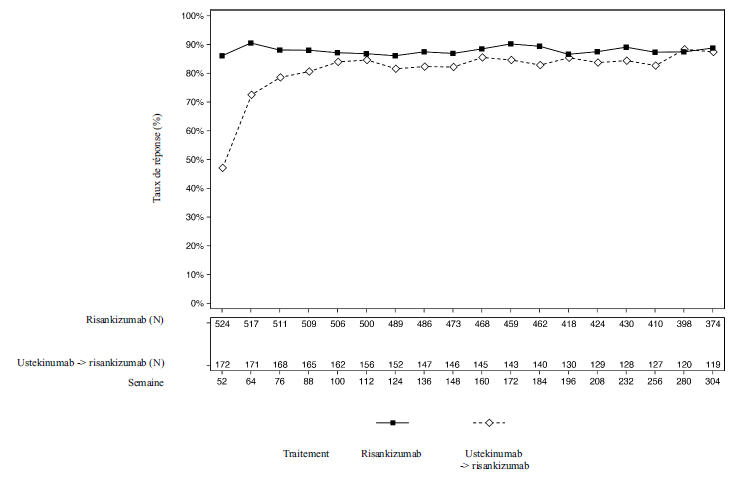
Figure 3 : Pourcentage de patients ayant obtenu une réponse de score sPGA « blanchi » ou « minime » dans LIMMITLESS, par visite (cas observés)
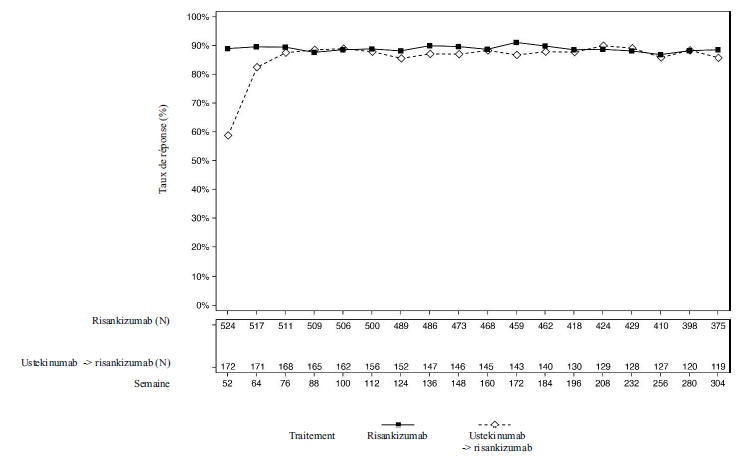
Les améliorations du score Dermatology Life Quality Index (DLQI 0 ou 1) ont été maintenues chez les patients recevant un traitement par risankizumab continu jusqu'à la semaine304 dans l'étude d'extension en ouvert LIMMITLESS.
Le profil de tolérance du risankizumab avec plus de 5 ans d'exposition était similaire à celui observé à 16 semaines.
IMMHANCE
L'étude IMMHANCE a inclus 507 patients (407 patients randomisés dans le groupe risankizumab 150 mg et 100 dans le groupe placebo). Les patients ont reçu le traitement à la semaine 0, à la semaine 4, puis toutes les 12 semaines. Les patients ayant été initialement randomisés dans le groupe risankizumab et ayant obtenu un sPGA « blanchi » ou « minime » à la semaine 28 ont été de nouveau randomisés pour recevoir le risankizumab toutes les 12 semaines jusqu'à la semaine 88 (avec un suivi de 16 semaines après l'administration de la dernière dose de risankizumab) ou pour arrêter le traitement.
À la semaine 16, le risankizumab s'est montré supérieur au placebo sur les deux co-critères principaux d'évaluation, à savoir un score sPGA « blanchi » ou « minime » (83,5 % dans le groupe risankizumab vs 7,0 % dans le groupe placebo) et un score PASI 90 (73,2 % dans le groupe risankizumab vs 2,0 % dans le groupe placebo).
Sur les 31 patients de l'étude IMMHANCE atteints d'une tuberculose (TB) latente et n'ayant pas reçu de traitement prophylactique durant l'étude, aucun n'a développé de TB active au cours de la période de suivi d'une durée moyenne de 55 semaines sous risankizumab.
Parmi les patients ayant obtenu un score sPGA « blanchi » ou « minime » à la semaine 28 dans IMMHANCE, cette réponse était maintenue à la semaine 104 chez 81,1 % (90/111) des patients rerandomisés pour poursuivre le traitement par risankizumab comparé à 7,1 % (16/225) de ceux re-randomisés pour arrêter le traitement par risankizumab. Parmi ces patients, 63,1 % (70/111) des patients re-randomisés pour poursuivre le traitement par risankizumab ont obtenu un score PGA « blanchi » à la semaine 104 comparé à 2,2 % (5/225) de ceux re-randomisés pour arrêter le traitement par risankizumab.
Parmi les patients ayant obtenu un score sPGA « blanchi » ou « minime » à la semaine 28 et dont le score est revenu à un niveau « modéré » ou « sévère » suite à l'arrêt du traitement par risankizumab, 83,7 % (128/153) ont de nouveau obtenu un score sPGA « blanchi » ou « minime » après 16 semaines de retraitement. Une perte du score sPGA « blanchi » ou « minime » était observée dès 12 semaines après un oubli de dose. Parmi les patients re-randomisés pour arrêter le traitement, 80,9 % (182/225) ont rechuté, et le délai médian de rechute était de 295 jours. Aucune caractéristique permettant de prédire le délai jusqu'à la perte de réponse ou la propension à présenter de nouveau une réponse n'a été identifiée au niveau individuel.
IMMVENT
L'étude IMMVENT a inclus 605 patients (301 patients randomisés dans le groupe risankizumab et
304 dans le groupe adalimumab). Les patients randomisés dans le groupe risankizumab ont reçu 150 mg du traitement à la semaine 0, à la semaine 4, puis toutes les 12 semaines. Les patients randomisés dans le groupe adalimumab ont reçu 80 mg à la semaine 0, 40 mg à la semaine 1, puis 40 mg une semaine sur deux jusqu'à la semaine 15. À partir de la semaine 16, les patients sous adalimumab ont poursuivi ou changé de traitement selon la réponse obtenue :
- réponse < PASI 50 : passage au traitement par risankizumab ;
- réponse PASI 50 à < PASI 90 : re-randomisation pour poursuivre l'adalimumab ou passer à risankizumab ;
- réponse PASI 90 : poursuite du traitement par adalimumab.
Les résultats obtenus sont présentés dans le Tableau 4.
Tableau 4 : Résultats d'efficacité et de qualité de vie à la semaine 16 chez des adultes atteints de psoriasis en plaques dans l'étude IMMVENT
|
|
Risankizumab (N = 301) n (%) |
Adalimumab (N = 304) n (%) |
|
sPGA « blanchi » ou « minime »a |
252 (83,7) |
183 (60,2) |
|
PASI 75 |
273 (90,7) |
218 (71,7) |
|
PASI 90a |
218 (72,4) |
144 (47,4) |
|
PASI 100 |
120 (39,9) |
70 (23,0) |
|
DLQI 0 ou 1b |
198 (65,8) |
148 (48,7) |
|
Toutes les comparaisons ont obtenu une valeur p < 0,001. a Co-critères principaux d'évaluation. b Pas d'impact sur la qualité de vie liée à la santé. |
|
|
Chez les patients sous adalimumab ayant obtenu un score PASI 50 à < PASI 90 à la semaine 16 et ayant été re-randomisés, les écarts entre les taux des réponses PASI 90 pour ceux passant au risankizumab et ceux poursuivant l'adalimumab ont été observés 4 semaines après la re-randomisation (respectivement 49,1 % vs 26,8 %).
Les résultats à 28 semaines après re-randomisation sont présentés dans le Tableau 5 et la Figure 4.
Tableau 5 : Résultats d'efficacité à 28 semaines après re-randomisation dans IMMVENT
|
|
Relais par risankizumab (N = 53) n (%) |
Poursuite avec adalimumab (N = 56) n (%) |
|
PASI 90 |
35 (66,0) |
12 (21,4) |
|
PASI 100 |
21 (39,6) |
4 (7,1) |
|
p < 0,001 pour toutes les comparaisons. |
|
|
Figure 4 : Évolution au cours du temps du score PASI 90 après re-randomisation dans IMMVENT
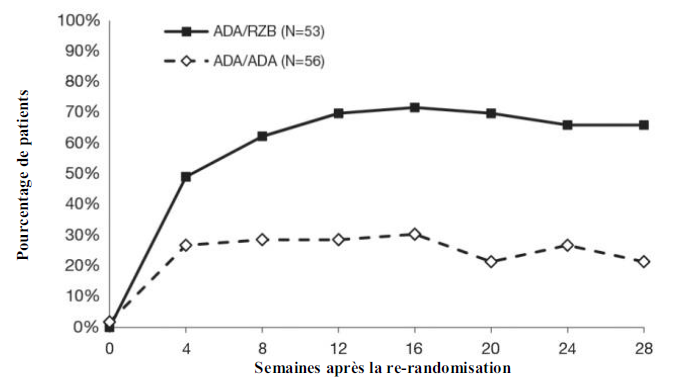
ADA/ADA : patients randomisés dans le groupe
adalimumab et poursuivant le traitement par adalimumab.
ADA/RZB : patients randomisés dans le groupe
adalimumab et passant au traitement par risankizumab.
p < 0,05 à la semaine 4, puis p < 0,001 à
chaque temps d'évaluation
à partir de la semaine 8.
Chez les 270 patients passés de l'adalimumab au risankizumab sans sevrage thérapeutique (wash-out), le profil de tolérance du risankizumab s'est avéré similaire à celui observé chez les patients ayant commencé le traitement par risankizumab après une période de sevrage thérapeutique de tous les traitements systémiques antérieurs.
Rhumatisme psoriasique
Il a été montré que le risankizumab améliore les signes et symptômes, la capacité fonctionnelle, la qualité de vie liée à la santé et la proportion de patients sans progression radiographique chez les adultes atteints de rhumatisme psoriasique (RP) actif.
L'efficacité et la tolérance du risankizumab ont été évaluées chez 1 407 patients atteints de RP actif au cours de 2 études randomisées en double aveugle contrôlées versus placebo (964 dans KEEPSAKE1 et 443 dans KEEPSAKE2).
Les patients inclus dans ces études présentaient un diagnostic de RP depuis au moins 6 mois selon les critères de classification du rhumatisme psoriasique (CASPAR), une durée médiane de RP de 4,9 ans à l'inclusion, ≥ 5 articulations douloureuses et ≥ 5 articulations gonflées, et un psoriasis en plaques actif ou un psoriasis unguéal à l'inclusion. 55,9 % des patients présentaient un psoriasis en plaques actif avec une surface corporelle atteinte (SCA) ≥ 3 %. Des enthésites et des dactylites étaient présentes chez respectivement 63,4 % et 27,9 % des patients. Dans l'étude KEEPSAKE1, au cours de laquelle le psoriasis unguéal a été plus spécifiquement évalué, 67,3 % des patients présentaient un psoriasis unguéal.
Dans les deux études, les patients ont été randomisés pour recevoir 150 mg de risankizumab ou un placebo aux semaines 0, 4 et 16. À partir de la semaine 28, tous les patients ont reçu du risankizumab toutes les 12 semaines.
Dans l'étude KEEPSAKE1, tous les patients présentaient des antécédents de réponse inadéquate ou d'intolérance à un traitement par DMARD non biologique et étaient naïfs de traitement biologique. Dans l'étude KEEPSAKE2, 53,5 % des patients présentaient des antécédents de réponse inadéquate ou d'intolérance à un traitement par DMARD non biologique et 46,5 % des patients présentaient des antécédents de réponse inadéquate ou d'intolérance à un traitement biologique.
Dans les deux études, 59,6 % des patients recevaient un traitement concomitant par méthotrexate (MTX), 11,6 % un traitement concomitant par DMARD non biologique autre que le MTX et 28,9 % le risankizumab en monothérapie.
Réponse clinique
Le traitement par risankizumab a entraîné une amélioration significative des indicateurs de l'activité de la maladie par rapport au placebo à la semaine 24. Dans les deux études, le critère principal d'évaluation était la proportion de patients ayant obtenu une réponse ACR20 selon les critères de l'American College of Rheumatology à la semaine24. Les principaux résultats d'efficacité sont présentés dans le Tableau 6.
Tableau 6. Résultats d'efficacité dans les études KEEPSAKE1 et KEEPSAKE2
|
|
KEEPSAKE1 |
KEEPSAKE2 |
||
|
Critère d'évaluation |
Placebo N = 481 n (%) |
Risankizumab N = 483 n (%) |
Placebo N = 219 n (%) |
Risankizumab N = 224 n (%) |
|
Réponse ACR20 |
||||
|
Semaine 16 |
161 (33,4) |
272 (56,3) a |
55 (25,3) |
108 (48,3) a |
|
Semaine 24 |
161 (33,5) |
277 (57,3) a |
58 (26,5) |
115 (51,3) a |
|
Semaine 52* |
- |
338/433 (78,1) |
- |
131/191 (68,6) |
|
Réponse ACR50 |
||||
|
Semaine 24 |
54 (11,3) |
162 (33,4) b |
20 (9,3) |
59 (26,3) b |
|
Semaine 52* |
- |
209/435 (48,0) |
- |
72/192 (37,5) |
|
Réponse ACR70 |
||||
|
Semaine 24 |
23 (4,7) |
74 (15,3) b |
13 (5,9) |
27 (12,0) c |
|
Semaine 52* |
- |
125/437 (28,6) |
- |
37/192 (19,3) |
|
Résolution des enthésites (LEI = 0) |
||||
|
Semaine 24* |
156/448 (34,8) d |
215/444 (48,4) a, d |
- |
- |
|
Semaine 52* |
- |
244/393 (62,1) d |
- |
- |
|
Résolution des dactylites (LDI = 0) |
||||
|
Semaine 24* |
104/204 (51,0) e |
128/188 (68,1) a, e |
- |
- |
|
Semaine 52* |
- |
143/171 (83,6) e |
- |
- |
|
Réponse MDA (activité minimale de la maladie) |
||||
|
Semaine 24 |
49 (10,2) |
121 (25,0) a |
25 (11,4) |
57 (25,6) a |
|
Semaine 52* |
- |
183/444 (41,2) |
- |
61/197 (31,0) |
|
*Patients pour lesquels les données sont disponibles, données présentées sous la forme n/N observé (%). a) Valeur p ≤ 0,001 avec contrôle de multiplicité, comparaison risankizumab versus placebo. b) Valeur p nominale ≤ 0,001, comparaison risankizumab versus placebo. c) Valeur p nominale ≤ 0,05, comparaison risankizumab versus placebo. d) Synthèse d'après des données poolées des études KEEPSAKE1 et KEEPSAKE2 pour les patients avec une valeur LEI > 0 à l'inclusion. e) Synthèse d'après des données poolées des études KEEPSAKE1 et KEEPSAKE2 pour les patients avec une valeur LDI > 0 à l'inclusion. |
||||
Réponse au cours du temps
Dans l'étude KEEPSAKE1, une réponse ACR20 plus importante a été observée dans le groupe risankizumab par rapport au groupe placebo dès la semaine 4 (25,7 %) et la différence en fonction du traitement s'est maintenue dans le temps jusqu'à la semaine 24 (Figure 5).
Figure 5. Pourcentage de patients ayant obtenu une réponse ACR20 dans l'étude KEEPSAKE1 jusqu'à la semaine 24
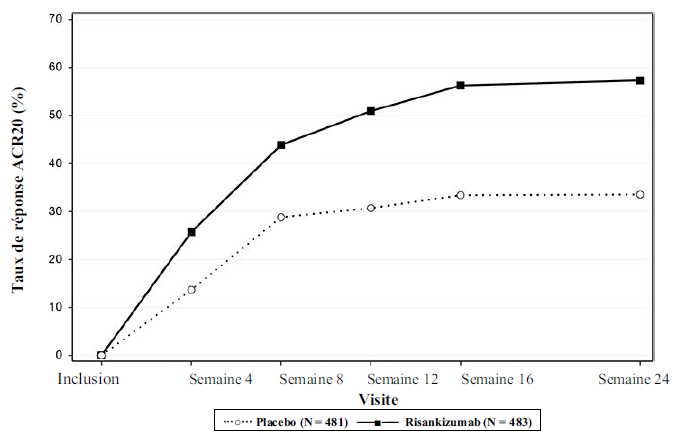
Une réponse ACR20 plus importante avec le risankizumab versus placebo a été observée dès la semaine 4 chez 19,6 % des patients dans l'étude KEEPSAKE2.
Les réponses observées dans les groupes risankizumab étaient similaires indépendamment de l'utilisation de DMARD non biologiques, du nombre de DMARD non biologiques antérieurs, de l'âge, du sexe, de l'origine ethnique et de l'IMC. Dans l'étude KEEPSAKE2, les réponses observées étaient indépendantes du traitement biologique antérieur.
Le profil de tolérance du risankizumab jusqu'à 52semaines d'exposition était similaire à celui observé jusqu'à 24 semaines.
Dans les deux études, la proportion de patients ayant obtenu une réponse basée sur le score PsARC modifié à la semaine 24 était plus élevée chez les patients recevant le risankizumab que chez ceux recevant le placebo. En outre, à la semaine 24, les patients recevant le risankizumab avaient obtenu une amélioration plus importante du score de l'activité de la maladie (28articulations) mesuré à l'aide de la CRP (DAS28-CRP) par rapport aux patients recevant le placebo. Les améliorations des scores PsARC et DAS28-CRP ont été maintenues jusqu'à la semaine 52.
Le traitement par risankizumab a permis une amélioration de chaque composante de l'ACR, de l'indice d'évaluation de la capacité fonctionnelle (HAQ-DI), de l'évaluation de la douleur et de la protéine C réactive ultrasensible (hsCRP) par rapport au placebo.
Le traitement par risankizumab a permis une amélioration statistiquement significative des manifestations cutanées du psoriasis chez les patients atteints de RP.
Le traitement par risankizumab a permis une amélioration statistiquement significative de l'indice modifié de sévérité du psoriasis unguéal (mNAPSI) et du score d'évaluation globale par le médecin de la sévérité du psoriasis des ongles et des mains sur une échelle de 5 points (PGA-F) chez les patients atteints de psoriasis unguéal à l'inclusion (67,3%) dans l'étude KEEPSAKE1. Cette amélioration s'est maintenue jusqu'à la semaine 52 (voir Tableau 7).
Tableau 7. Résultats d'efficacité dans le psoriasis unguéal dans l'étude KEEPSAKE1
|
|
Placebo N = 338 |
Risankizumab N = 309 |
|
Variation du score mNAPSI par rapport à l'inclusion a |
||
|
Semaine 24 |
-5,57 |
-9,76 b |
|
Semaine 52 |
- |
-13,64 |
|
Variation du score PGA-F par rapport à l'inclusion a |
||
|
Semaine 24 |
-0,4 |
-0,8 b |
|
Semaine 52 |
- |
-1,2 |
|
PGA-F blanchi/minimal et amélioration de grade ≥ 2 c |
||
|
Semaine 24 n (%) |
30 (15,9) |
71 (37,8) d |
|
Semaine 52 n (%) |
- |
105 (58,0) |
|
a) Synthèse pour les patients atteints de psoriasis unguéal à l'inclusion (placebo N = 338 ; risankizumab N = 309 ; à la semaine 52, pour mNAPSI, risankizumab N (observé) = 290, pour PGA-F, risankizumab N (observé) = 291). b) Valeur p ≤ 0,001 avec contrôle de multiplicité, comparaison risankizumab versus placebo. c) Synthèse pour les patients atteints de psoriasis unguéal et présentant un score d'évaluation globale PGA-F « léger », « modéré » ou « sévère » à l'inclusion (placebo N = 190 ; risankizumab N = 188, à la semaine 52 risankizumab N (observé) = 181). d) Valeur p nominale ≤ 0,001, comparaison risankizumab versus placebo. |
||
Réponse radiographique
Dans l'étude KEEPSAKE1, l'inhibition de la progression des lésions structurales a été évaluée par radiographie et exprimée en termes de variation du score total de Sharp modifié (mTSS) à la semaine 24, par rapport à l'inclusion. Le score mTSS a été modifié pour le RP en ajoutant les articulations interphalangiennes distales (IPD) de la main. À la semaine 24, la progression moyenne des lésions structurales avec le risankizumab (score mTSS moyen 0,23) par rapport au placebo (score mTSS moyen 0,32) n'était pas statistiquement significative. À la semaine 24, la proportion de patients ne présentant pas de progression radiographique (définie comme une modification du score mTSS par rapport à l'inclusion≤ 0) était plus importante avec le risankizumab (92,4 %) par rapport au placebo (87,7 %). Cette réponse s'est maintenue jusqu'à la semaine 52.
Capacité fonctionnelle et qualité de vie liée à la santé
Dans les deux études, les patients traités par risankizumab ont montré une amélioration statistiquement significative de la capacité fonctionnelle par rapport à l'inclusion, évaluée par le score HAQ-DI à la semaine 24 (KEEPSAKE1 (-0,31) par rapport aux patients recevant le placebo (-0,11) (p ≤ 0,001)), (KEEPSAKE2 (-0,22) par rapport aux patients recevant le placebo (-0,05) (p ≤ 0,001)). À la semaine 24, une plus grande proportion de patients avaient obtenu une diminution cliniquement significative d'au moins 0,35 du score HAQ-DI par rapport à l'inclusion, dans le groupe risankizumab par rapport au placebo. Les améliorations de la capacité fonctionnelle ont été maintenues jusqu'à la semaine 52.
Dans les deux études, les patients traités par risankizumab présentaient des améliorations significatives des valeurs des composantes physiques du score SF-36 V2 et du score FACIT-fatigue à la semaine 24 par rapport aux patients recevant le placebo, avec des améliorations maintenues jusqu'à la semaine 52.
À l'inclusion, une atteinte axiale a été signalée chez 19,6 % (7,9 % diagnostiquée par radiographie ou IRM) des patients de l'étude KEEPSAKE1 et 19,6 % (5 % diagnostiquée par radiographie ou IRM) des patients de l'étude KEEPSAKE2. Les patients ayant une atteinte axiale cliniquement évaluée traités par risankizumab ont montré des améliorations par rapport à l'inclusion de l'indice d'activité de la spondylarthrite ankylosante (BASDAI) à la semaine 24 par rapport aux patients recevant le placebo. Les améliorations ont été maintenues jusqu'à la semaine 52. En raison du faible nombre de patients étudiés, les preuves de l'efficacité du risankizumab chez les patients souffrant de RP avec atteinte axiale confirmée par radiographie ou IRM, sont insuffisantes.
Population pédiatrique
L'Agence européenne des médicaments a différé l'obligation de soumettre les résultats d'études réalisées avec Skyrizi dans un ou plusieurs sous-groupes de la population pédiatrique dans le traitement du psoriasis en plaques et du rhumatisme psoriasique (voir rubrique Posologie et mode d'administration pour les informations concernant l'usage pédiatrique).
La pharmacocinétique du risankizumab était similaire chez les patients atteints de psoriasis en plaques et les patients atteints de rhumatisme psoriasique.
Absorption
Le profil pharmacocinétique du risankizumab s'est avéré linéaire avec une augmentation de l'exposition proportionnelle à la dose dans les intervalles de dose allant de 18 mg à 300 mg et de 0,25 mg/kg à 1 mg/kg en administration sous-cutanée, et de 200 mg à 1200 mg et 0,01 mg/kg à 5 mg/kg en administration intraveineuse.
Après administration du risankizumab par voie sous-cutanée, les concentrations plasmatiques maximales ont été atteintes en 3 à 14 jours avec une biodisponibilité absolue estimée à 89 %. Lors de l'administration d'une dose de 150 mg à la semaine 0, à la semaine 4, puis toutes les 12 semaines, les concentrations plasmatiques maximale et minimale à l'état d'équilibre estimées ont été respectivement de 12 µg/mL et 2 µg/mL.
La bioéquivalence entre une injection unique de risankizumab 150 mg et deux injections de risankizumab 75 mg en seringue préremplie a été démontrée. La bioéquivalence entre le risankizumab 150 mg solution injectable en seringue préremplie et en stylo prérempli a également été démontrée.
Distribution
La valeur moyenne (± écart-type) du volume de distribution à l'état d'équilibre (Vee) du risankizumab était de 11,4 (± 2,7) L dans les études de phase III menées chez des patients atteints de psoriasis, ce qui montre que la distribution du risankizumab est essentiellement confinée aux espaces interstitiels et vasculaires.
Biotransformation
Les anticorps monoclonaux thérapeutiques IgG sont généralement dégradés en petits peptides et acides aminés par des voies cataboliques de la même façon que les IgG endogènes. Il n'est pas attendu que le risankizumab soit métabolisé par les enzymes du cytochrome CYP450.
Élimination
La clairance (CL) systémique moyenne (± écart-type) du risankizumab était de 0,3 L/jour (± 0,1) dans les études de phase III menées chez des patients atteints de psoriasis. La demi-vie d'élimination terminale moyenne du risankizumab était de 28 à 29 jours dans les études de phase III menées chez des patients atteints de psoriasis.
En tant qu'anticorps monoclonal IgG1, le risankizumab ne devrait pas faire l'objet d'une filtration glomérulaire rénale ni d'une excrétion sous forme inchangée dans les urines.
Linéarité/non-linéarité
Le profil pharmacocinétique du risankizumab s'est avéré linéaire avec des augmentations approximativement dose-proportionnelles en matière d'exposition systémique (Cmax et ASC) dans les intervalles de dose évalués, à savoir 18 à 300 mg ou 0,25 à 1 mg/kg en administration sous-cutanée, chez des volontaires sains ou des patients atteints de psoriasis.
Interactions
Une étude d'interaction a été menée chez des patients atteints de psoriasis en plaques pour évaluer l'effet de l'administration répétée du risankizumab sur la pharmacocinétique de substrats tests sensibles du cytochrome P450 (CYP). Après administration du traitement par risankizumab, les expositions à la caféine (substrat du CYP1A2), à la warfarine (substrat du CYP2C9), à l'oméprazole (substrat du CYP2C19), au métoprolol (substrat du CYP2D6) et au midazolam (substrat du CYP3A) étaient comparables aux valeurs observées avant le traitement par risankizumab, ce qui indique l'absence d'interactions cliniquement significatives avec ces enzymes.
Les analyses pharmacocinétiques de population ont montré que l'exposition au risankizumab n'était pas altérée par les traitements concomitants utilisés par certains patients atteints de psoriasis en plaques ou de rhumatisme psoriasique et inclus dans les études cliniques.
Populations particulières
Population pédiatrique
La pharmacocinétique du risankizumab n'a pas été établie chez les patients pédiatriques.
Personnes âgées
Sur l'effectif de 2 234 patients atteints de psoriasis en plaques et exposés à risankizumab, 243 étaient âgés de 65 ans et plus et 24 de 75 ans et plus. Sur 1 542 patients atteints de rhumatisme psoriasique et exposés au risankizumab, 246 étaient âgés de 65 ans et plus et 34 patients de 75 ans et plus. Dans l'ensemble, il n'y a pas de différence observée dans l'exposition au risankizumab entre les patients âgés et les patients plus jeunes qui recevaient risankizumab.
Insuffisants rénaux ou hépatiques
Aucune étude spécifique n'a été menée pour déterminer l'effet de l'insuffisance rénale ou hépatique sur la pharmacocinétique du risankizumab. Selon les analyses pharmacocinétiques de population, les taux de créatinine sérique, la clairance de la créatinine et les marqueurs de la fonction hépatique (ALAT/ASAT/bilirubine) n'avaient pas d'effet significatif sur la clairance du risankizumab chez les patients atteints de psoriasis en plaques ou de rhumatisme psoriasique.
En tant qu'anticorps monoclonal IgG1, le risankizumab est essentiellement éliminé par catabolisme intracellulaire et ne devrait pas être métabolisé par les enzymes hépatiques du cytochrome P450 ni éliminé par voie rénale.
Poids
La clairance du risankizumab ainsi que son volume de distribution augmentent avec l'augmentation du poids, ce qui peut induire une efficacité réduite chez les patients ayant un poids élevé (> 130 kg).
Cependant, cette observation est basée sur un nombre limité de sujets. Il n'y a actuellement pas d'adaptation posologique recommandée en fonction du poids.
Sexe et origine ethnique
La clairance du risankizumab n'était pas significativement influencée par le sexe ou l'origine ethnique chez les patients adultes atteints de psoriasis en plaques ou de rhumatisme psoriasique. Lors d'une étude de pharmacocinétique clinique chez des volontaires sains, aucune différence cliniquement significative de l'exposition au risankizumab n'a été observée chez les sujets chinois ou japonais, comparativement aux sujets caucasiens.
Le risankizumab n'a aucun effet ou un effet négligeable sur l'aptitude à conduire des véhicules et à utiliser des machines.
Les données non cliniques issues des études de toxicologie en administration répétée, incluant des études de pharmacologie de sécurité et une étude de toxicologie approfondie sur le développement pré- et post natal chez les singes cynomolgus à des doses allant jusqu'à 50 mg/kg/semaine (induisant des expositions d'environ 70 fois l'exposition clinique à la dose maximale recommandée chez l'homme [DMRH]), n'ont pas révélé de risque particulier pour l'homme.
Aucune étude de mutagénèse ou de cancérogénèse n'a été réalisée avec le risankizumab. Au cours d'une étude de toxicité chronique de 26 semaines chez les singes cynomolgus à des doses allant jusqu'à 50 mg/kg/semaine (soit environ 70 fois l'exposition clinique à la DMRH), aucune lésion prénéoplasique ou néoplasique, ni aucun effet indésirable immunotoxique ou de nature cardiovasculaire n'a été observé.
Skyrizi 150 mg, solution injectable en stylo prérempli
Avant l'injection, les patients doivent sortir la boîte du réfrigérateur et la laisser revenir à température ambiante à l'abri de la lumière directe du soleil (pendant 30 à 90 minutes) sans retirer le stylo prérempli de la boîte.
La solution doit être incolore à jaune et limpide à légèrement opalescente.
Skyrizi 150 mg, solution injectable en seringue préremplie
Avant l'injection, les patients peuvent sortir la boîte du réfrigérateur et la laisser revenir à température ambiante à l'abri de la lumière directe du soleil (pendant 15 à 30 minutes) sans retirer la seringue préremplie de la boîte.
La solution doit être incolore à jaune et limpide à légèrement opalescente.
Skyrizi 75 mg, solution injectable en seringue préremplie
Avant l'injection, les patients peuvent sortir la boîte du réfrigérateur et la laisser revenir à température ambiante à l'abri de la lumière directe du soleil (pendant 15 à 30 minutes) sans retirer les seringues préremplies de la boîte.
La solution doit être incolore à légèrement jaune et limpide à légèrement opalescente.
Le contenu des deux seringues préremplies doit être injecté afin d'administrer la dose complète de 150 mg.
Précautions particulières générales
Avant toute utilisation, un examen visuel de chaque stylo prérempli ou seringue préremplie est recommandé. La solution peut contenir quelques particules de produit, translucides à blanches. Skyrizi ne doit pas être utilisé si la solution est trouble, présente une couleur anormale ou contient de grosses particules. Ne pas agiter le stylo prérempli ou la seringue préremplie.
Les instructions complètes d'utilisation sont fournies dans la notice.
Chaque stylo prérempli ou seringue préremplie est à usage unique exclusivement.
Tout médicament non utilisé ou déchet doit être éliminé conformément à la réglementation en vigueur.
Liste I
Prescription réservée aux spécialistes et services DERMATOLOGIE
Prescription réservée aux spécialistes et services MEDECINE INTERNE
Prescription réservée aux spécialistes et services RHUMATOLOGIE
Médicament d'exception.
Solution injectable
La solution est incolore à jaune et limpide à légèrement opalescente.
Seringue préremplie en verre intégrée dans un stylo prérempli avec un manchon automatique de protection de l'aiguille.
Skyrizi 150 mg est disponible en boîte contenant 1 stylo prérempli.
Chaque stylo prérempli contient 150 mg de risankizumab dans 1 mL de solution.
Le risankizumab est un anticorps monoclonal humanisé de type immunoglobuline G1 (IgG1) produit par des cellules ovariennes de hamster chinois par la technique de l'ADN recombinant.
Pour la liste complète des excipients, voir rubrique Liste des excipients.
Acétate de sodium trihydraté
Acide acétique
Tréhalose dihydraté
Polysorbate 20
Eau pour préparations injectables
Acide acétique
Tréhalose dihydraté
Polysorbate 20
Eau pour préparations injectables